Research
Medicinal chemistry
Design and synthesis of anti-viral agents, structure-activity relationships
One of our main interests is the design and synthesis of anti-viral agents that inhibit the function of certain viral proteins and the exploration of their structure-activity relationships. Current targets are hepatitis B (HBV), influenza A and hepatitis C (HCV) viruses.
- Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles
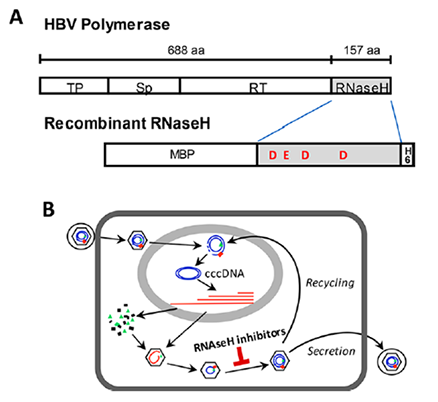
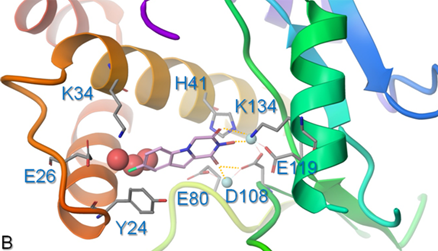
- Hepatitis B virus (HBV) chronically infects >250 million people and kills nearly a million annually, and current antivirals cannot clear the infection or adequately suppress disease. The virus replicates by reverse transcription, and the dominant antiviral drugs are nucleos(t)ide analogs that target the viral reverse transcriptase. We are developing antivirals targeting the other essential viral enzymatic activity, the ribonuclease H (RNaseH).
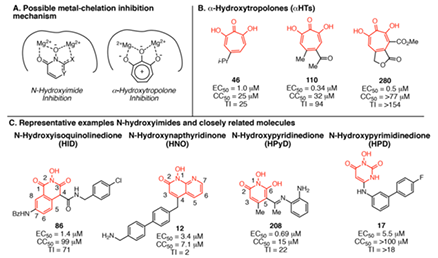
- HBV RNaseH inhibitors with nanomolar efficacies against viral replication in culture have been identified in the α-hydroxytropolone, N-hydroxyisoquinolinedione, N-hydroxypyridinedions, and N-hydroxynapthyridinone chemotypes. We are a Lab that focuses on RNaseH inhibitors, current synthetic approaches, and challenges to optimizing the inhibitors into leads for clinical assessment.
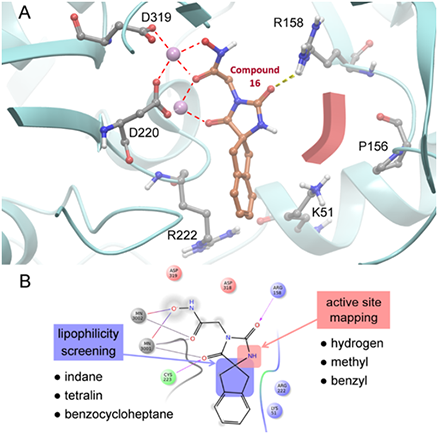
- Novel flutimide analogues targeting the influenza virus PA endonuclease
- Influenza is a highly contagious infectious disease that affects millions of people every year. In the twentieth century, influenza caused more fatalities in Europe than any other infectious disease. Current vaccines against influenza virus require annual updating due to continuous variation of the viral antigens. Thus, anti-influenza virus drugs are vital as a first line of defense. At present, two classes of antivirals are available: the neuraminidase inhibitors oseltamivir and zanamivir, and the M2 proton channel blockers amantadine and rimantadine. For both drug classes, viral resistance is an emerging concern and, hence, novel antiviral agents with an alternative mode of action and favorable resistance profile are urgently needed. A promising new target is the influenza virus PA endonuclease, which performs the "cap-snatching" reaction during viral mRNA synthesis, and is essential for virus replication. The crystal structure of the N-terminal part of PA (PA-Nter) containing the catalytic endonuclease domain, was recently revealed, enabling structure-based development of PA inhibitors. Our group synthesized and evaluated several analogues of the natural compound Flutimide (a fungus-derived influenza virus endonuclease inhibitor with a 2,6-diketo-Δ3-piperazine motif). Using an enzymatic PA endonuclease activity assay, we show that these compounds block the enzymatic activity of PA-Nter. Theoretical calculations are also undertaken for providing a structural rationale for the interaction between the synthesized analogues and the viral protein and a number of molecular interactions contributing to binding affinity and specificity were elucidated. Theoretical results are supported by biochemical analyses of the enzymatic activity inhibition. Overall, our data reveal exciting strategies for the design and optimization of novel influenza virus inhibitors that target the viral PA endonucleaser.
- New synthetic aminoadamantane compounds
- We are also interested in new aminoadamantane compounds with potent anti-influenza virus A activity, bearing high selectivity indices (see list of publications). The reference compounds in this series, amantadine and rimantadine are current anti-influenza virus A drugs. Their protonated form blocks the influenza A M2 ion channel protein activity via interactions of the drug with the transmembrane domain of the tetrameric M2 protein, that is, the 25 residue M2TM peptide tetramer (M2TM sequence of Udorn H3N2 strain is: SSDPLVVAASIIGILHLILWILDRL).
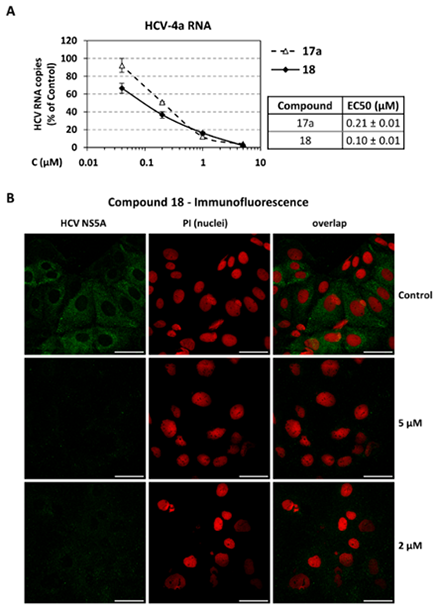
- Scaffold Hybridization Strategy towards potent hydroxamate-based inhibitors of Flaviviridae viruses and Trypanosoma species
- Infections with Flaviviridae viruses, such as hepatitis C virus (HCV) and dengue virus (DENV) pose global health threats. Infected individuals are at risk of developing chronic liver failure or haemorrhagic fever respectively, often with a fatal outcome if left untreated. Diseases caused by tropical parasites of the Trypanosoma species, T. brucei and T. cruzi, constitute significant socioeconomic burden in sub-Saharan Africa and continental Latin America, yet drug development is under-funded. Anti-HCV chemotherapy is associated with severe side effects and high cost, while Dengue has no clinically approved therapy and antiparasitic drugs are outdated and difficult to administer. Moreover, drug resistance is an emerging concern. Consequently, the need for new revolutionary chemotherapies is urgent. By utilizing a molecular framework combination approach, we combined two distinct chemical entities with proven antiviral and trypanocidal activity into a novel hybrid scaffold attached by an acetohydroxamic acid group (CH2CONHOH), aiming at derivatives with dual activity. The novel spiro-carbocyclic substituted hydantoin analogues were rationally designed, synthesized and evaluated for their potency against three HCV genotypes (1b, 3a, 4a), DENV and two Trypanosoma species (T. brucei, T. cruzi). They exhibited significant EC50 values and remarkable selectivity indices. Several modifications were undertaken to further explore the structure activity relationships (SARs) and confirm the pivotal role of the acetohydroxamic acid metal binding group.
Design and Synthesis of Antiparasitic Agents, Structure-Activity Relationships
Human African trypanosomiasis (HAT), is caused by tsetse fly transmitted parasites of the Trypanosoma brucei species complex. Over 50 million people in sub-Saharan Africa are at risk. In 2009, there were 30,000 cases, although during epidemics, this level can increase >10-fold. Chagas disease (or American trypanosomiasis), is caused by Trypanosoma cruzi and affects 8-10 million people in Latin America, resulting in >15,000 deaths per year. Therapy for trypanosomal infections is unsatisfactory due to toxic side effects, the development of resistance, and in many cases, the need for parenteral administration. With no immediate prospect of vaccines, there is a great need to develop new antitrypanosome agents with an acceptable efficacy and safety profile.
Based on our previous results on structure activity relationship (SAR) of trypanocidal agents we synthesize derivatives with a variety of structural modification at different positions of our lead compound, in order to determine how structural features affect the trypanocidal activity of compounds and improve their ADME, toxicological and pharmacokinetic properties (see list of publications).
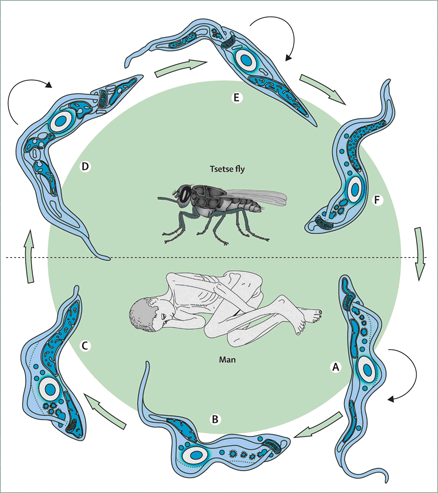
Figure: Vector and parasite.
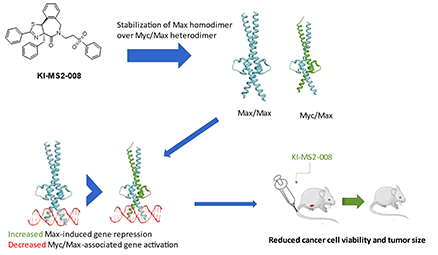
Cancer comprises a heterogeneous disease, characterized by diverse features such as constitutive expression of oncogenes and/or downregulation of tumor suppressor genes. MYC constitutes a master transcriptional regulator, involved in many cellular functions and is aberrantly expressed in more than 70% of human cancers. The Myc protein belongs to a family of transcription factors whose structural pattern is referred to as basic helix-loop-helix-leucine zipper. Myc binds to its partner, a smaller protein called Max, forming an Myc:Max heterodimeric complex that interacts with specific DNA recognition sequences (E-boxes) and regulates the expression of downstream target genes. Myc protein plays a fundamental role for the life of a cell, as it is involved in many physiological functions such as proliferation, growth and development since it controls the expression of a very large percentage of genes (~15%). However, despite the strict control of MYC expression in normal cells, MYC is often deregulated in cancer, exhibiting a key role in stimulating oncogenic process affecting features such as aberrant proliferation, differentiation, angiogenesis, genomic instability and oncogenic transformation.
Our group aims to meticulously describe the fundamental role of MYC in tumorigenesis and highlight its importance as an anticancer drug target. We focus mainly on the design and synthesis of different categories of novel small molecules that act as inhibitors of Myc function in diverse ways hence offering great opportunities for an efficient cancer therapy. This knowledge will provide significant information for the development of novel Myc inhibitors and assist to the design of treatments that would effectively act against Myc-dependent cancers.