Research Areas
1. Medicinal chemistry - Ion Channel Blockers
A. Design and Synthesis of anti-viral agents, structure-activity relationships
One of our main interests is the design and synthesis of anti-viral agents that inhibit the function of certain viral ion channel proteins and the exploration of their structure-activity relationships. Our main target is the influenza A virus but herpes simplex viruses are also of interest.
-
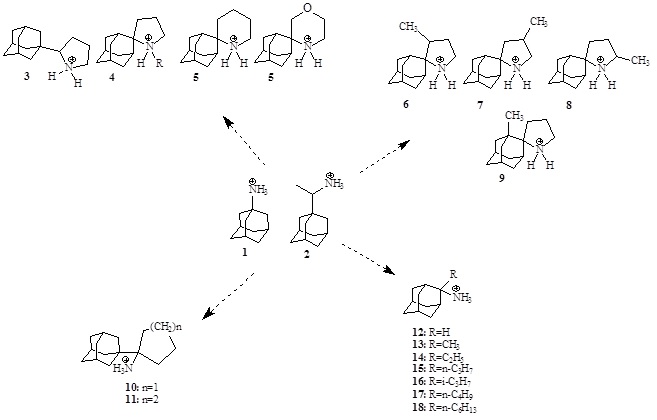
We have synthesized aminoadamantane (Aamt) derivatives with potent anti-influenza virus A activity, some of them bearing high selectivity indices (Scheme 1). The prototype compounds in this series, amantadine (Amt) (1) and (Rim) rimantadine (2) were one of the two classes of commercial anti-influenza virus A drugs. Since 2005, 1/2-resistant mutants have become prevalent globally, abrogating clinical usefulness of 1 and 2. The protonated form of the drug inhibits the influenza WT-A/M2 membrane ion channel protein activity which is necessary for virus replication and budding. This is realized by blocking the pore of the transmembrane domain of the tetrameric wild type A/M2 protein, that is, the 25 residue Α/M2TM peptide tetramer (the M2TM peptide of Udorn strain includes the sequence SSDPLVVAASIIGILHLILWILDRL).
-
The use of 1 and 2 drugs has been limited because of the rapid emergence of resistant mutants including amino acid changes in the M2TM sequence, from which S31N is dominant. We have found Amt analogues with sizeable adducts active against the 2009 H1N1 swine flu pandemic contained AM2 S31N mutation being active without blocking full M2 protein. We also synthesize new conjugates of Amt with polar heads which are active against WSN/33 virus (an S31N virus) considered to be a target S31N virus. We investigate also new Aamt derivatives with activity against WSN/33 viruses bearing the most important mutations for human infections (V27A, L26F).
-
Currently in collaboration we are preparing complexes of Aamt derivatives with metals against influenza to investigate a possible boost in activity through a refined ligand that may block directly or through a synergistic effect more efficiently channel pore.
Scheme 1. Structure evolution of some aminoadamantanes synthesized in our lab.
The application of whole cell antiviral testing assays is essential for establishing biological activity (performed in collaboration with other groups). However, investigation of the interaction between Amt analogues and A/M2TM is prerequisite for understanding interactions at the molecular level and making subsequent suggestions using structure-based drug design methods (see B below).
Relevant references
- Stylianakis, I.; Kolocouris, A.; Kolocouris, N.; Fytas, G.; Foscolos, G. B.; Padalko, E.; Neyts, J.; De Clercq, E. et al. Spiro[pyrrolidine-2,2-adamantanes]: Synthesis, Anti-Influenza Virus Activity and Conformational Properties. Biοorg. Med. Chem. Letters 2003, 13, 1699-1703. http://dx.doi.org/10.1016/S0960-894X(03)00231-2
- Tataridis, G.; Fytas, A.; Kolocouris, A.; Fytas, C.; Kolocouris, N.; Foscolos, G. B.; Padalko, E.; Neyts, J.; De Clercq, E. Influence of an Additional 2-Αmino Substituent to the 1-Aminoethyl Pharmacophore Group on the Potency of Rimantadine Against Influenza Virus. Biοorg. Med. Chem. Letters 2007, 17, 692-696. http://dx.doi.org/10.1016/j.bmcl.2006.10.092
- Kolocouris, A.; Spearpoint, P.; Martin, S. R.; Hay, A. J.; López-Querol, M.; Sureda, F. X.; Padalko, E.; Jeyts, J.; De Clercq, E. Comparisons of the Influenza Virus A M2 Channel Binding Affinities, Anti-Influenza Virus Potencies and NMDA Antagonistic Activities of 2-Alkyl-2-Aminoadamantanes and Analogues. Biοorg. Med. Chem. Letters 2008, 18, 6156-6160. http://dx.doi.org/10.1016/j.bmcl.2008.10.003
- Fytas, C.; Kolocouris, A.; Fytas, G.; Zoidis, G.; Valmas, C.; Basler, C. F. Influence of an Additional Amino Group on the Potency of Aminoadamantanes Against Influenza Virus A. II – Synthesis of Spiropiperazines and In-Vitro Activity Against Influenza A H3N2 Virus. Bioorg. Chem. 2010, 38, 247-251. http://dx.doi.org/10.1016/j.bioorg.2010.09.001
- Kolocouris, A.; Tzitzoglaki, C.; Johnson, F. B.; Zell, R.; Wright, A. K.; Cross, T. A.; Tietjen, I.; Fedida, D.; Busath, D. D. Aminoadamantanes with Persistent In Vitro Efficacy Against H1N1 (2009) Influenza A. J. Med. Chem. 2014, 57, 4629-39. http://pubs.acs.org/doi/abs/10.1021/jm500598u
- Drakopoulos, A.; Ma, C.; Freudenberger, K.; Hoffmann, A.; Gauglitz, G.; Schmidtke, M.; Wang, J.; Kolocouris, A. A case-study considering the development of anti-influenza aminoadamantanes: alchemical free energy calculations, binding affinities, electrophysiology blocking studies and in-vitro potency. In preparation.
- Tzitzoglaki, C.; Drakopoulos, A.; Mazzanti, A.; Stylianakis, I.; Kolocouris, A. Approaches to primary tert-alkyl amines. In preparation.
B. Interactions between M2 protein blocker drugs and their receptor
A second research area, which is complementary to the first, is aiming a) at understanding and inspecting the interactions between Amt analogues and influenza M2TM-tetramer receptor using various techniques like NMR spectroscopy b) at investigation of the structure of the system c) the design of new more effective inhibitors using computational methods.
i. Experimental work
- Measurement of binding constants of Aamt derivatives against the full M2 in acidic pH using an assay based on the inhibition of Trp41 fluorescence quenching upon drug binding at acidic pH (inhibition of quenching is effected by His37 protonation which must cause a conformational change) induced by the Aamt ligand binding in the M2 channel pore.
- Measurement of binding constants of Aamt derivatives to the transmembrane domain M2TM at alkaline pH using Isothermal Titration Calorimetry (ITC). The binding constants values are used to comprehend the results of the calculations.
- Measurement of the blocking effect of Aamt derivatives against the full M2 protein.
- Studying the point-mutations on A/M2TM sequence caused by the interaction of Aamt derivatives with influenza A virus.
- The synthesis of labeled reporter molecules (ligands or peptide receptor fragments) is realized. The peptides are synthesized under optimized solid phase peptide synthesis conditions. NMR signals of the reporter molecules are used to follow the ligand-receptor binding.
- We have already succeed in following the ligand-Α/M2TM receptor binding using simple 1H NMR spectroscopy of a fluorine-labeled Amt ligand.
- We have applied 19F NMR spectroscopy to study the complex of Amt with a labeled Α/M2TM (19F-Trp-M2TM). At pH 8, when the ligand binds to the receptor, 19F-Trp signals from both the free and bound states of the Α/M2TM tetramer are resolved (Figure 1). Thus, the differentiation of bound and unbound states of the Α/M2TM receptor by 19F NMR may provide a system for SAR studies, We plan to work on a SAR study using 19F NMR spectroscopy.
- We investigate the interaction of Aamt derivatives with M2TM in phospholipid bilayers using ssNMR and small-angle X-ray scattering rays of membrane systems (SAXS) in collaboration with other groups.
- We investigate the structure of M2TM - ligand complexes using X-ray crystallography in collaboration with other groups.
Figure 1. Interaction between a fluorine labeled Amt analog (F-Am) and the fluorine tryptophane-labeled influenza A/M2TM receptor peptide (F-M2TM) probed by 19F NMR spectroscopy of the receptor

Relevant references
- binding constants of ligands against full M2 protein.
- Eleftheratos, S.; Ortore, G.; Kolocouris, A.; Martinelli, A.; Spearpoint, P.; Martin, S.; Hay, A. Interaction of Aminoadamantane Derivatives with the Influenza A Virus M2 Channel - Docking Using a Pore Blocking Model. Bioorganic and Medicinal Chemistry Letters 2010, 20, 4182-4187. http://dx.doi.org/10.1016/j.bmcl.2010.05.049
- Gkeka, P.; Eleftheratos, S.; Kolocouris, A.; Cournia, Z. Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Analogs as Blockers of the Influenza M2 Channel. J. Chem. Theory Comput. 2013, 9, 1272-81. http://pubs.acs.org/doi/abs/10.1021/ct300899n.
- binding constants of ligands against M2TM.
- Homeyer, N.; Ioannidis, H.; Kolarov, F.; Gauglitz, G.; Zikos, C.; Kolocouris, A.; Gohlke, H. Interpreting Thermodynamic Profiles of Aminoadamantane Compounds Inhibiting the M2 Proton Channel of Influenza A by Free Energy Calculations. J. Chem. Inf. Model. 2016, 56, 110-26. http://pubs.acs.org/doi/abs/10.1021/acs.jcim.5b00467
- Ioannidis, H.; Drakopoulos, A.; Tzitzoglaki, C.; Homeyer, N.; Kolarov, F.; Gkeka, P.; Freudenberger, K.-M.; Liolios, C. C.; Gauglitz, G.; Cournia, Z.; Gohlke, H.; Kolocouris, A. Alchemical Free Energy Calculations and Isothermal Titration Calorimetry Measurements of Aminoadamantanes Bound to the Closed State of Influenza A/M2TM. J. Chem. Inf. Model. Just Accepted Manuscript. http://pubs.acs.org/doi/abs/10.1021/acs.jcim.6b00079
- Drakopoulos, A.; Ma, C.; Freudenberger, K.; Hoffmann, A.; Gauglitz, G.; Schmidtke, M.; Wang, J.; Kolocouris, A. A case-study considering the development of anti-influenza aminoadamantanes: alchemical free energy calculations, binding affinities, electrophysiology blocking studies and in-vitro potency. In preparation.
- electrophysiology.
- Kolocouris, A.; Tzitzoglaki, C.; Johnson, F. B.; Zell, R.; Wright, A. K.; Cross, T. A.; Tietjen, I.; Fedida, D.; Busath, D. D. Aminoadamantanes with Persistent In Vitro Efficacy Against H1N1 (2009) Influenza A. J. Med. Chem. 2014, 57, 4629-39. http://pubs.acs.org/doi/abs/10.1021/jm500598u
- Tzitzoglaki, C.; Wright, A.; Freudenberger, K.; Kolarov, F.; Tietjen, I.; Fedida, D.; Gauglitz, G.; Cross, T. A.; Kolocouris, A. Aminoadamantanes Bound to Influenza WT and S31N M2TM - Evidence for weak binding - no blocking Inhibition of Proton Conductance of S31N M2 Protein. Submitted.
- Drakopoulos, A.; Ma, C.; Freudenberger, K.; Hoffmann, A.; Gauglitz, G.; Schmidtke, M.; Wang, J.; Kolocouris, A. A case-study considering the development of anti-influenza aminoadamantanes: alchemical free energy calculations, binding affinities, electrophysiology blocking studies and in-vitro potency. In preparation.
- solution NMR.
- Kolocouris, A.; Broadhurst, R. W.; Hansen, R. Interaction Between an Amantadine Analog and the Transmembrane Portion of the Influenza A M2 Protein in Liposomes Probed by 1H NMR Spectroscopy of the Ligand. Journal of Medicinal Chemistry 2004, 47, 4975-4978. http://dx.doi.org/10.1021/jm0496685
- Kolocouris, A.; Zikos, C.; Broadhurst, R. W. 19F NMR Trapping of the Complex Between Amantadine and the Receptor Portion of the Influenza A M2 Ion Channel in DPC micelles. Biοorganic and Medicinal Chemistry Letters 2007, 17, 3799-3813. http://dx.doi.org/10.1016/j.bmcl.2007.04.100
- ssNMR.
- Kolocouris, A.; Tzitzoglaki, C.; Johnson, F. B.; Zell, R.; Wright, A. K.; Cross, T. A.; Tietjen, I.; Fedida, D.; Busath, D. D. Aminoadamantanes with Persistent In Vitro Efficacy Against H1N1 (2009) Influenza A. J. Med. Chem. 2014, 57, 4629-39. http://pubs.acs.org/doi/abs/10.1021/jm500598u
- Tzitzoglaki, C.; Wright, A.; Freudenberger, K.; Kolarov, F.; Tietjen, I.; Fedida, D.; Gauglitz, G.; Cross, T. A.; Kolocouris, A. Aminoadamantanes Bound to Influenza WT and S31N M2TM - Evidence for weak binding - no blocking Inhibition of Proton Conductance of S31N M2 Protein. Submitted.
ii. Calculations of relative free energies of binding
-
We have measured the binding affinities of amino-adamantane derivatives: a) at acidic pH against the full length M2 protein using the inhibition of quenching of Trp41 fluorescence at acidic pH induced by the Aamt ligand binding (this research included the expression and purification of the full M2 protein) b) at alkaline pH against tetrameric M2TM corresponding to the sequence of different influenza A strains (Udorn, Weybbridge, G34A etc) using ITC (this research requires the solid phase peptide synthesis and purification of the highly lipophilic M2TM 25-residues peptides). The binding constants are used to guide the interpretation of the calculations predictions.
- We are performing FEP/MD alchemical calculations to test the prediction of relative binding free energies of several Amt analogues to M2TM tetramer at both acidic and alkaline pH. Free energy perturbation (FEP) methods are widely recognized as the most accurate for calculating binding free energy differences. FEP method is based on statistical thermodynamics and has an accuracy in the order of 1 kcal/mol on the relative binding free energy prediction, starting with an accurate geometric model and full sampling. We plan to test rigorous free energy calculations based on Thermodynamic Integration (TI).
- Starting structures are produced by docking calculations.
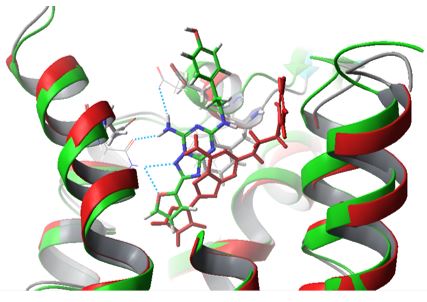
- During the past 3 years, there has been a huge surge to characterize M2TMUdorn (M2TMWT) structurally through solution state NMR, crystallography, and, most relevant with regard to protein surroundings, ssNMR. Thus, extensive experimental investigation have been published for Amt- and Rim-M2TMUdorn complex. However, the complex model should be refined when a mutant M2TM bound to different Aamt derivatives is studied (for example Weybridge or S31N). In that case starting models are produced using MD simulations.
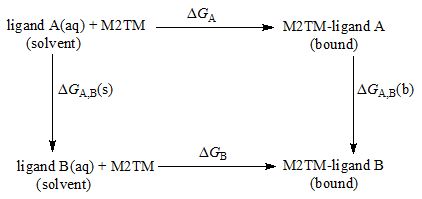
Scheme 2. Thermodynamic cycle for the calculation of relative free energies of binding applied to alchemical tranformations of Aamt derivatives. ΔGA, ΔGB are the free energies of transfer of A and B from the aqueous phase (unbound state) to the bound state, respectively. ΔGA,B(s) and ΔGA,B(b) are the free energy differences of the mutation of A into B in aqueous solution, and bound to the protein respectively calculated using the BAR method.
The calculations are based on the thermodynamic cycle of Scheme 2 and on the following equation:.
ΔΔGligand A→ B = ΔGA,B(b) - ΔGA,B(s)
The method predicts nicely relative binding affinities with good to high correlation and the results are encouraging for design new molecules.
- Most recently in collaboration, we interpreted thermodynamic profiles of ITC for Aamt compounds binding to M2TM by MM-PBSA calculations and used this information for successfully and prospectively prioritizing Aamt derivatives.
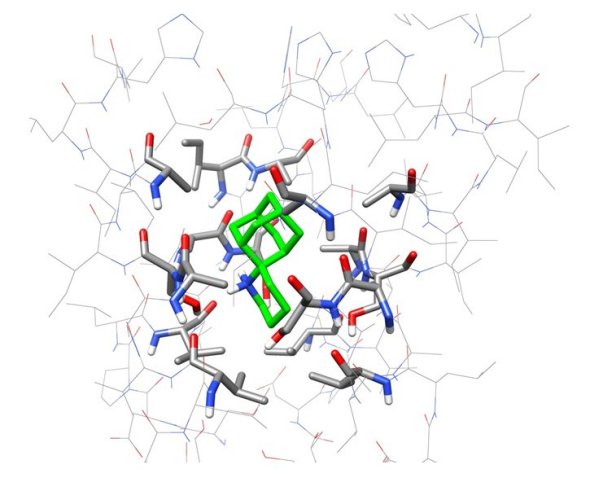
Figure 2. Representative snapshot from the simulation of spiro[pyrrolidine-2,2'-adamantane] bound to A/M2TM. Ten waters are shown between the ligand and His37 residues. Two hydrogen bonds between the ammonium group of the ligand and two water molecules are shown. Hydrogen bonding together with van der Waals interactions of the adamantane core with Val27 and Ala30 stabilize the ligand inside the pore with its ammonium group oriented towards the C-end.
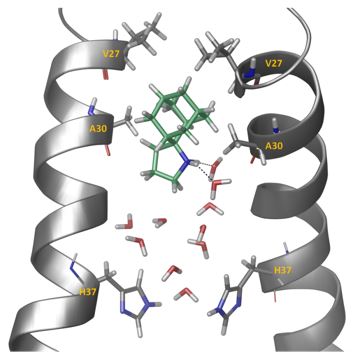
Figure 3. Relative binding free energies computed by the BAR approach (ΔΔGBAR) plotted against relative binding free energies derived from ITC data (ΔΔGexperiment) for M2TMUdorn embedded in a DMPC bilayer. The correlation line obtained by a linear least-squares fit including all data points are shown (solid, black line). Maximal errors in ΔΔGBAR and ΔΔGexperiment are shown as error bars along the vertical and horizontal axes, respectively. In the panel the square correlation coefficient (R2), the p value, and the slope (s) of the least-squares fit line is included.

Relevant references
- Eleftheratos, S.; Ortore, G.; Kolocouris, A.; Martinelli, A.; Spearpoint, P.; Martin, S.; Hay, A. Interaction of Aminoadamantane Derivatives with the Influenza A Virus M2 Channel - Docking Using a Pore Blocking Model. Bioorganic and Medicinal Chemistry Letters 2010, 20, 4182-4187. http://dx.doi.org/10.1016/j.bmcl.2010.05.049
- Gkeka, P.; Eleftheratos, S.; Kolocouris, A.; Cournia, Z. Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Analogs as Blockers of the Influenza M2 Channel. J. Chem. Theory Comput. 2013, 9, 1272-81. http://pubs.acs.org/doi/abs/10.1021/ct300899n
- Gleed, M. L.; Ioannidis, H.; Kolocouris, A.; Busath, D. D. Resistance-Mutation (N31) Effects on Drug Orientation and Channel Hydration in Amantadine-Bound Influenza A M2. J. Phys. Chem. B. 2015, 119, 11548-59. http://www.ncbi.nlm.nih.gov/pubmed/26268449
- Homeyer, N.; Ioannidis, H.; Kolarov, F.; Gauglitz, G.; Zikos, C.; Kolocouris, A.; Gohlke, H. Interpreting Thermodynamic Profiles of Aminoadamantane Compounds Inhibiting the M2 Proton Channel of Influenza A by Free Energy Calculations. J. Chem. Inf. Model. 2016, 56, 110-26. http://pubs.acs.org/doi/abs/10.1021/acs.jcim.5b00467
- Ioannidis, H.; Drakopoulos, A.; Tzitzoglaki, C.; Homeyer, N.; Kolarov, F.; Gkeka, P.; Freudenberger, K.-M.; Liolios, C. C.; Gauglitz, G.; Cournia, Z.; Gohlke, H.; Kolocouris, A. Alchemical Free Energy Calculations and Isothermal Titration Calorimetry Measurements of Aminoadamantanes Bound to the Closed State of Influenza A/M2TM. J. Chem. Inf. Model. Just Accepted Manuscript. http://pubs.acs.org/doi/abs/10.1021/acs.jcim.6b00079
- Drakopoulos, A.; Ma, C.; Freudenberger, K.; Hoffmann, A.; Gauglitz, G.; Schmidtke, M.; Wang, J.; Kolocouris, A. A case-study considering the development of anti-influenza aminoadamantanes: alchemical free energy calculations, binding affinities, electrophysiology blocking studies and in-vitro potency. In preparation.
2. Medicinal chemistry - Virtual screening
Most conventional programs perform docking calculations and provide an estimation of the energy of interaction for the ligand-receptor complex. The energy constituents are van der Waals energy, Coulomb interaction, H-bonding interaction etc. The programs miss to include a critical term, that is, the desolvation penalty for transferring the ligand from solution to the receptor area and this energy cost can be really important. Thus, a ligand A may have a stronger interaction energy with a receptor with respect to a ligand B, but can be a weaker binder if the tendency of A to stay in solution is greater to that of B, that is, A has to pay a higher desolvation penalty for moving from solution to the receptor.
Consider the thermodynamic cycle of Scheme 3.
Scheme 3. Thermodynamic cycle used for the calculation of the free energy of binding of ligand A to receptor R

The free energy for the formation of ligand-receptor complex can be calculated using the end-points of this thermodynamic cycle, that is, the bound and unbound states of the ligand. Thus, the method includes two simulations (MD or Monte Carlo), one for the solvated receptor-drug complex and one for the ligand in solution. In both cases the interaction of the ligand with the environment (recepror in the bound state or solvent in the unbound state) is analyzed into a van der Waals constituent and to an electrostatic constituent. The free energy of binding is calculated for the simulation ensembles according to equation.
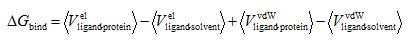
The method is difficult to be applied for screening a compounds libraries by applying MD or MC simulations with explicit consideration of water molecules due to the high computational cost.
A solution to that problem is the calculation of the solvent effects using an implicit salvation model (instead of the computationally expensive explicit representation) which is based on the numerical solution of PB-equation (PBEQ calculations). The MM-PBSA or MM-GBSA model is based on that concept and the binding energy is calculated according to equation
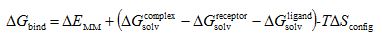
The calculation of the free energies for transferring ligand, protein receptor and complex from the gas to the solution phase (free energies of solvation, ΔGsolv) is needed according to the cycle of Scheme 4. The electrostatic component is calculated using the Poisson theory or the generalized Born model (Generalized Born) which is computationally more efficient than Poisson theory· the non-polar component of the free energy of solvation is calculated based on the surface of the cave formed by solute inside the solvent. ΔΕΜΜ is calculated in the gas phase divided to Coulomb and van der Waals interaction energy. Entropy change often is taken to be zero.
A significant reduction of computational resources is realized when MD or MC simulation is replaced by a geometry minimization.
A virtual screening of a small compounds library against adenosine receptor is under investigation. After docking and scoring with GOLD/ChemPLP and rescoring with ROCS we applied CHARMM/PBEQ for post-scoring taking into account the desolvation penalty. We discovered a series of low micromolar inhibitors with A3AR selectivity.
Figure 4. Close-up view of a representative snapshot from a 70 ns MD simulation of ZM241385 inside A2AAR in phospholipid bilayer.
Relevant references
- Lagarias, P.; Convertino, M.; Ortore, O.; Klotz, K.-N.; Kolocouris, A. Structure-Guided of Novel Adenosine Receptor Ligands With Subtype A3 Selectivity. In preparation.
3. Medicinal chemistry - Molecular dynamics simulations
Current projects include the application of MD simulations of membrane protein systems embedded into various membrane models to investigate: a) the binding and selectivity of aminoadamantane ligands inside influlenza A M2TM pore and its mutants; b) the binding and selectivity of ligands against adenosine receptor subtypes and c) the binding of various antihypertensive drugs (AT1-receptor antagonists) against angiotensin receptor.
4. Conformational analysis using Dynamic NMR spectroscopy – Weak intramolecular interactions
A. Dynamic NMR spectroscopy
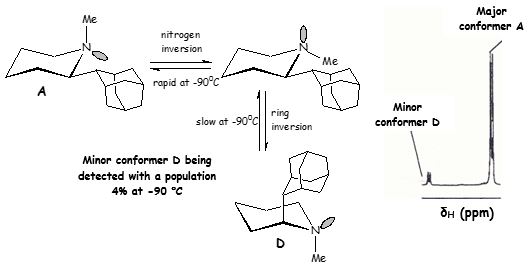
We have a special interest in the past with the field of solution conformational analysis of synthetic organic molecules with restricted internal motion (ring inversion, nitrogen inversion, bond rotation) using Dynamic NMR spectroscopy. The distinct NMR signals of the populated conformers which can be observed usually only at low temperatures can be identified using a combination of 2D NMR experiments (COSY, HMQC, HMBC, NOESY, EXSY) and computational chemistry (molecular mechanics or ab initio calculations). Using such a detailed experimental analysis, minor conformers can be identified which are probably important for the biological potency profile of a compound.
- We have already addressed the Dynamic NMR analysis of some anti-influenza virus A aminoadamantane derivatives and intending to run experiments with new analogues.
Scheme 4. Populated conformers A, D of the anti-influenza A 2-(2-adamantyl)-N-methylpiperidine being identified at -90 0C when ring inversion became a slow process at the NMR time scale
Relevant references
- Kolocouris, A.; Mikros, E.; Kolocouris, N. Stereodynamics of Ring and Nitrogen Inversion in Spiroheterocycles. Conformational Analysis of N-methylspiro[morpholine 3,2 adamantane] and N-methylspiro [piperidine 2,2 adamantane] Using NMR Spectroscopy and Theoretical Calculations. J. Chem. Soc., Perkin Trans. 2, 1998, 1701-1708. http://dx.doi.org/10.1039/a705868c
- Kolocouris, A.; Outeiriño, J. G.; Anderson, J. E.; Fytas, G.; Foscolos, G. B.; Kolocouris, N. The effect of Neighbouring 1- and 2-adamantyl group substitution on the conformations and stereodynamics of N-methylpiperidine. Dynamic NMR spectroscopy and molecular mechanics calculations. J. Org. Chem. 2001, 66, 4989-4997. http://dx.doi.org/10.1021/jo0016677
- Stylianakis, I.; Kolocouris, A.; Kolocouris, N.; Fytas, G.; Foscolos, G. B.; Padalko, E.; Neyts, J.; De Clercq, E. et al. Spiro[pyrrolidine-2,2-adamantanes]: Synthesis, Anti-Influenza Virus Activity and Conformational Properties. Biοorg. Med. Chem. Letters 2003, 13, 1699-1703. http://dx.doi.org/10.1016/S0960-894X(03)00231-2
- Kolocouris, A. The Effect of Spirοadamantane Substitution on the Conformational Preferences of N-Me Pyrrolidine and N-Me Piperidine: A Description Based on Dynamic NMR Spectroscopy and Ab Initio Correlated Calculations. Tetrahedron 2009, 65, 9428-9435. http://dx.doi.org/10.1016/j.tet.2009.08.071
- Tzitzoglaki, C.; Drakopoulos, A.; Mazzanti, A.; Stylianakis, I.; Kolocouris, A. Approaches to primary tert-alkyl amines. In preparation.
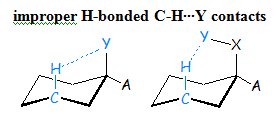
B. Weak intramolecular interactions – Improper Hydrogen-Bonded C-H∙∙∙Y contacts
A second research project is aiming at exploring intramolecular and intermolecular C-H∙∙∙Y contacts and their possible contribution to the thermodynamic stability of the system under study.
- We applied MP2 and various DFT functionals, NBO calculations and QTAIM analysis to propose for the first time that the C-Hax∙∙∙Yax-C contacts in the axial cyclohexane derivatives are not just Pauli repulsive in nature but also include attractive dispersion interactions, and may be characterized even as improper hydrogen-bonding component in certain cases.
Scheme 5. C-Hax∙∙∙Yax contacts.
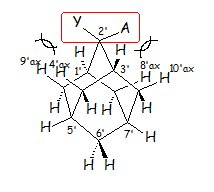
- We have published an elegant way to investigate the features of the 1H NMR spectrum of an axially substituted cyclohexane ring, through studying the rigid cyclohexane ring sub-units included in a suitable substituted adamantane derivative (Schemes 5, 6). The full assignment of the spectra was realized by applying GIAO calculations.
- We have been recently succeed in building and tuning the strength of cyclohexane improper hydrogen bonded C-Hax∙∙∙Yax-C contacts using adamantane molecules as templates (Schemes 5, 6). A relation between spectral parameters and strength of these contacts was observed.
- A current project investigates the significance of intramolecular and intermolecular C-H∙∙∙Y-C and C-H∙∙∙H-C contacts on the stabilization of certain protein structures.
Scheme 6. In 2,2-disubstituted adamantanes each of the two substituents Y, A adopts an axial or equatorial orientation in a different cyclohexane ring sub-unit of adamantane.
Relevant references
- Kolocouris, A. Ranking the effect of [1A(ax), 1B(eq)] vs [1A(eq), 1B(ax)] cyclohexane ring substitution on the 1H chemical shifts of γ-methylene cyclohexane ring protons using 2,2-disubstituted adamantanes as models. Tet. Lett. 2007, 48, 2117-2122. http://dx.doi.org/10.1016/j.tetlet.2007.01.121
- Κolocouris, A. C-Hax∙∙∙Yax Contacts in Cyclohexane Derivatives Revisited – Identification of Improper Hydrogen Bonded Contacts. J. Org. Chem. 2009, 74, 1842-1849. http://pubs.acs.org/doi/full/10.1021/jo801835a
- Zervos, N.; Kolocouris, A. Improper hydrogen-bonded cyclohexane C–Hax···Yax contacts: experimental evidence from 1H NMR spectroscopy of suitable axial cyclohexane models. Tet. Lett. 2010, 51, 2453-2456. http://dx.doi.org/10.1016/j.tetlet.2010.02.170
- Kolocouris, A.; Zervos, N.; De Proft, F.; Koch, A. Improper hydrogen bonded cyclohexane C-Hax···Yax contacts: theoretical predictions and experimental evidence from 1H NMR spectroscopy of suitable axial cyclohexane models. J. Org. Chem. 2011, 76, 4432-4443. http://pubs.acs.org/doi/abs/10.1021/jo102353f
- Kolocouris, A.; Koch, A.; Kleinpeter, E.; Stylianakis, I. 2-Substituted and 2,2-disubstituted adamantane derivatives as models for studying substituent chemical shifts and C–Hax⋯Yax cyclohexane contacts—results from experimental and theoretical NMR spectroscopic chemical shifts and DFT structures. Tetrahedron 2015, 71, 2463–2481. http://www.sciencedirect.com/science/article/pii/S0040402015000873
- Silva Lopez, C.; Fazza, O. N.; De Proft, F.; Kolocouris, A. Are the cyclohexane C-Hax···Yax contacts purely repulsive? A combined NBO, QTAIM and DFT Computational Analysis in a Selected Test Sample of Monosubstituted Cyclohexanes. In Preparation.
5. Computational chemistry
We are interesting in applying structure calculations and molecular simulations to address problems related to the structure, conformation and medicinal chemistry of organic compounds - biomolecules. Current projects include as was described above:
- Docking calculations of ligands library to the M2TM tetramer receptor.
- The application of ab initio computations and ONIOM methodology (for example mixed ab-initio/AM1 calculations) to investigate the stability of model α-helical peptides and the influenza virus A M2TM α-helical peptides.
- ONIOM calculations on the complex between M2TM and aminoadamantane ligands (mixed ab-initio/AM1 or ab-initio/MM calculations) using the best selected poses from the docking calculations as starting structures.
- Calculations of relative free energies of binding, MD simulations.
- The investigation of weak hydrogen bonded C-Hax∙∙∙Yax contacts in cyclohexane models and lipophilic a-helices using ab-initio calculations, NBO and QTAIM analysis.
- The assignment of the carbon chemical shifts in 2,2-disubstituted adamantanes using GIAO calculations
- The evaluation of various computational chemistry methods (molecular mechanics calculations, ab-initio QM methods) on their ability to predict the experimental conformational preferences of organic molecules and drugs.
Figure 5. Structure of the peptide monomer of the M2TM influenza virus A receptor of aminoadamantane drugs using mixed B3LYP/AM1 calculations (ONIOM method)
Ac-VVAASIGILHLILWILDRL-NH2 (Udorn strain)
Figure 6. Partition of an aminoadamantane drug M2TM complex for high level calculations of the binding site and low level of the surrounding system using the ONIOM method
and recently:
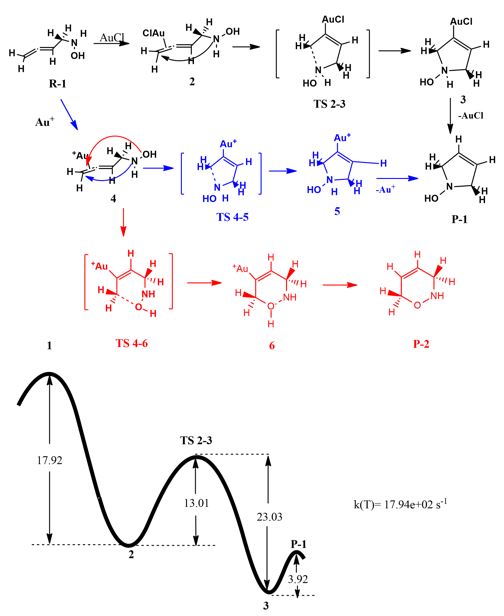
- The analysis of transition metal catalyzed mechanisms using DFT methods and appropriate algorithms for transition states investigation in the potential energy surface.
Scheme 7. Computational study of gold-catalyzed cycloisomerization of functionalized allenes.
Relevant references
- Eleftheratos, S.; Ortore, G.; Kolocouris, A.; Martinelli, A.; Spearpoint, P.; Martin, S.; Hay, A. Interaction of Aminoadamantane Derivatives with the Influenza A Virus M2 Channel - Docking Using a Pore Blocking Model. Bioorganic and Medicinal Chemistry Letters 2010, 20, 4182-4187. http://dx.doi.org/10.1016/j.bmcl.2010.05.049
- Gkeka, P.; Eleftheratos, S.; Kolocouris, A.; Cournia, Z. Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Analogs as Blockers of the Influenza M2 Channel. J. Chem. Theory Comput. 2013, 9, 1272-81. http://pubs.acs.org/doi/abs/10.1021/ct300899n
- Gleed, M. L.; Ioannidis, H.; Kolocouris, A.; Busath, D. D. Resistance-Mutation (N31) Effects on Drug Orientation and Channel Hydration in Amantadine-Bound Influenza A M2. J. Phys. Chem. B. 2015, 119, 11548-59. http://www.ncbi.nlm.nih.gov/pubmed/26268449
- Homeyer, N.; Ioannidis, H.; Kolarov, F.; Gauglitz, G.; Zikos, C.; Kolocouris, A.; Gohlke, H. Interpreting Thermodynamic Profiles of Aminoadamantane Compounds Inhibiting the M2 Proton Channel of Influenza A by Free Energy Calculations. J. Chem. Inf. Model. 2016, 56, 110-26. http://pubs.acs.org/doi/abs/10.1021/acs.jcim.5b00467
- Ioannidis, H.; Drakopoulos, A.; Tzitzoglaki, C.; Homeyer, N.; Kolarov, F.; Gkeka, P.; Freudenberger, K.-M.; Liolios, C. C.; Gauglitz, G.; Cournia, Z.; Gohlke, H.; Kolocouris, A. Alchemical Free Energy Calculations and Isothermal Titration Calorimetry Measurements of Aminoadamantanes Bound to the Closed State of Influenza A/M2TM. J. Chem. Inf. Model. Just Accepted Manuscript. http://pubs.acs.org/doi/abs/10.1021/acs.jcim.6b00079
- Kolocouris, A.; Mikros, E.; Kolocouris, N. Stereodynamics of Ring and Nitrogen Inversion in Spiroheterocycles. Conformational Analysis of N-methylspiro[morpholine3,2΄adamantane] and N-methylspiro [piperidine2,2΄adamantane] Using NMR Spectroscopy and Theoretical Calculations. J. Chem. Soc., Perkin Trans. 2, 1998, 1701-1708. http://dx.doi.org/10.1039/a705868c
- Kolocouris, A.; Outeiriño, J. G.; Anderson, J. E.; Fytas, G.; Foscolos, G. B.; Kolocouris, N. The effect of Neighbouring 1- and 2-adamantyl group substitution on the conformations and stereodynamics of N-methylpiperidine. Dynamic NMR spectroscopy and molecular mechanics calculations. J. Org. Chem. 2001, 66, 4989-4997. http://dx.doi.org/10.1021/jo0016677
- Kolocouris, A.; Zervos, N.; De Proft, F.; Koch, A. Improper hydrogen bonded cyclohexane C-Hax···Yax contacts: theoretical predictions and experimental evidence from 1H NMR spectroscopy of suitable axial cyclohexane models. J. Org. Chem. 2011, 76, 4432-4443. http://pubs.acs.org/doi/abs/10.1021/jo102353f
- Silva Lopez, C.; Fazza, O. N.; De Proft, F.; Kolocouris, A. Are the cyclohexane C-Hax···Yax contacts purely repulsive? A combined NBO, QTAIM and DFT Computational Analysis in a Selected Test Sample of Monosubstituted Cyclohexanes. In Preparation.
- Kolocouris, A. Evaluation of the Stability of Model a-Helical Peptides Using ab-initio Calculations, NBO and QTAIM analysis. Manuscript in preparation.
- Stylianakis, I.; Kolocouris, A. Testing the Performance of various Molecular Mechanics Force Fields on Predicting the Conformational Preferences in a Large Set of Small Organic Molecules and Drugs. Work in progress.
- Kyriakidi, S.; Kolocouris, A.; Silva Lopez, C. Computational Study of Gold-Catalyzed Cycloisomerization of Functionalized Allenes. In Preparation.
6. Former projects
A. Conformational analysis of molecules with high flexibility
In the case of highly complex and very flexible molecules the combination of NOESY experiments and molecular dynamics or Monte Carlo simulations of molecular trajectory can give valuable information for their average conformational preferences. Former projects included the conformational analysis of drug derivatives and bioactive peptides.
Figure 7. Superimposition of the low energy conformers of a bioactive peptide with its non-peptide drug antagonist; the peptide conformer structure was derived from a NOE constrained molecular dynamics simulation

Relevant references
- Mavromoustakos, T.; Kolocouris, A.; Zervou, M.; Roumelioti, P.; Matsoukas, J.; Weisemann, R. An Effort to Understand the Molecular Basis of Hypertension Through the Study of Conformational Analysis of Losartan and Sarmesin Using a Combination of NMR Spectroscopy and Theoretical Calculations. J. Med. Chem. 1999, 42, 1722-1729. http://dx.doi.org/10.1021/jm980499w
- Matsoukas, J.; Polayova, L.; Ancas, J.; Mavromoustakos, T.; Kolocouris, A.; Roumelioti, P.; Vlahakos, D. V.; Yamdagni, R.; Wu, Q.; Moore, G. J. The Design and Synthesis of a Potent Angiotensin II Cyclic Analogue Confirms the Ring Cluster Receptor Conformation of the Hormone Angiotensin II. Biοorg. Med. Chem. 2000, 8, 1-10. http://dx.doi.org/10.1016/S0968-0896(99)00266-7
- Mavromoustakos, T.; Calogeropoulou, T.; Koufaki, M; Kolocouris, A.; Daliani, I.; Demetzos, C.; Meng, Z.; Makriyannis, A.; Balzarini, J.; De Clercq, E. Εther Phospholipid-AZT Conjugates Possessing Anti-HIV and Antitumor Cell Activity. Synthesis, Conformational analysis, and Study of their Thermal Effects on Membrane Bilayers. J. Med. Chem. 2001, 44, 1702-1709. http://dx.doi.org/10.1021/jm001121c
B. Structure elucidation of bioactive molecules using NMR spectroscopy
In collaboration with natural product chemists we were involved in the structure elucidation, and relative stereochemistry verification of natural products using various 2D NMR spectroscopy techniques (COSY, HMQC, HMBC, NOESY).
Relevant references
- Kolocouris A.; Mavromoustakos T.; Demetzos C.; Terzis A.; Grdadolnik S.; Structure Εlucidation and Conformational Properties of a Novel Bioactive Clerodane Diterpene Using a Combination of High Field NMR Spectroscopy, Computational Analysis and Χ-ray Diffraction. Biοorganic and Medicinal Chemistry Letters 2000, 11, 837-840. http://dx.doi.org/10.1016/S0960-894X(01)00072-5
- Demetzos, C.; Kolocouris, A.; Anastasaki, T. A simple and rapid method for the differentiation of C-13 manoyl oxide epimers in biologically important samples using GC-MS analysis supported with NMR spectroscopy and computational chemistry results. Bioorg. Med. Chem. Lett. 2002, 12, 3605-9. http://www.sciencedirect.com/science/article/pii/S0960894X02007928
|